
This is an ongoing feature that will be updated daily until complete, check back often.
Anemia is found worldwide, in fact, according to the World Health Organization (WHO), about 25% of the world’s population suffers from some sort of anemia. While you may only encounter a few types as a Combat Medic in the US military, you may encounter several different types while deployed. Having a basic understanding may help you recognize it and save a life.
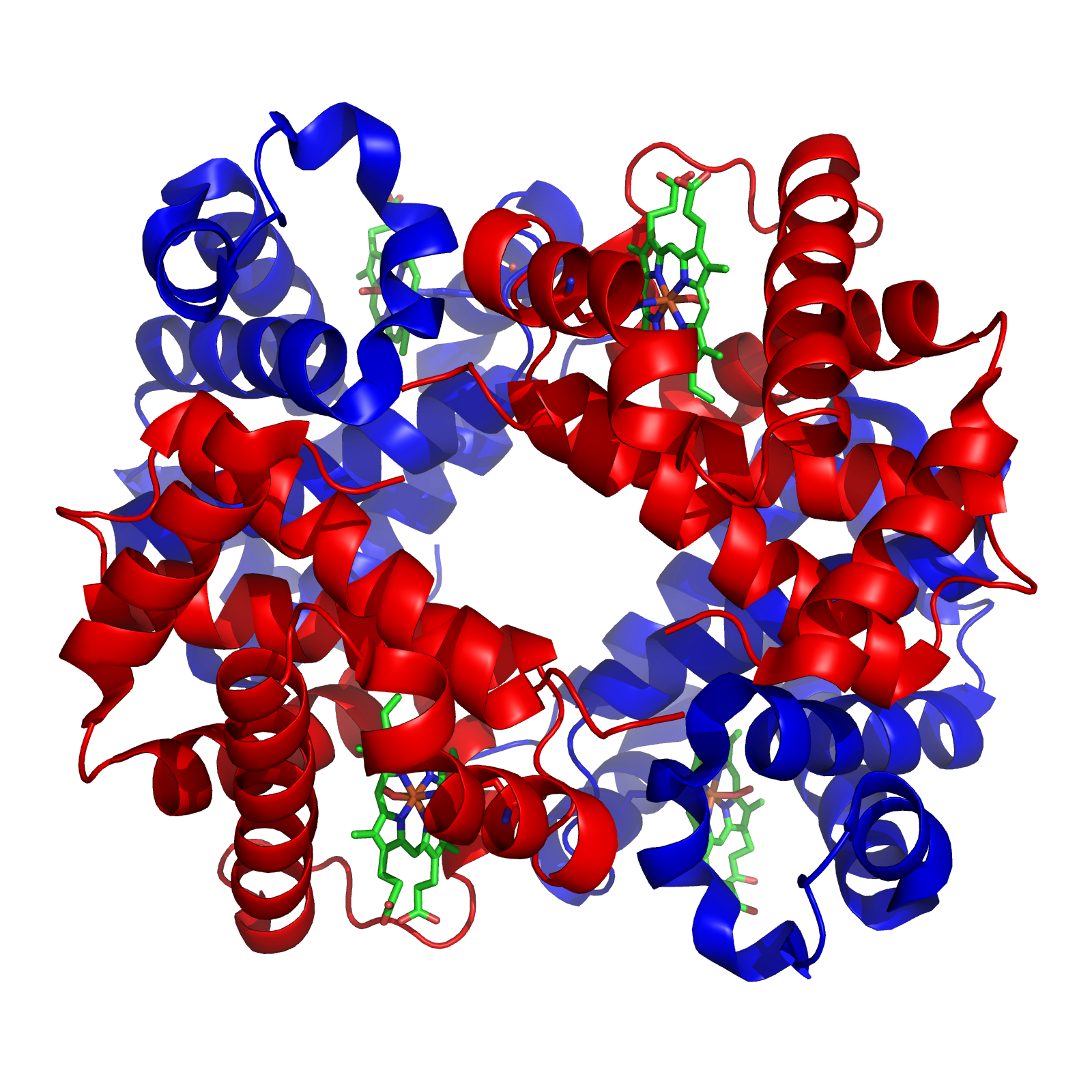
A normal RBC has a lot of hemoglobin. Normal hemoglobin has a tetramer of two α chains and two β chains, this is called hemoglobin A. There are others, but will be explained later. Hemoglobin is made of heme (iron + protoporphyrin) and a globin protein. The heme ring (see picture) has iron in the center, where oxygen is bound.
Classifications of Anemia
Anemias can be classified in a few ways.
- Problem making RBCs
- Intrinsic problems making RBCs
- Hemoglobinopathies-sickle cell disease
- Thalassemia
- RBC membrane defects-Hereditary Spherocytosis
- RBC enzyme defect-G6PD Deficiency
- Paroxysmal Nocturnal Hemoglobinuria-acquired disease
- Intrinsic problems making RBCs
- Destruction or loss of RBCs
- Extrinsic problems, hemolytic anemias
- Immunohemolytic anemia
- Microangiopathic hemolytic anemia-mechanical injury to RBC
- Parasites-Malaria
- Extrinsic problems, hemolytic anemias
Or we can classify them by the RBC size, which tends to be more common. RBC size is called Mean Corpuscular Volume (MCV). Normal is between 80-100 fL. High MCV indicates larger RBCs and lower MVCs indicate smaller RBCs.
- Microcytic (MCV <80 fL)
- Iron deficiency anemia (IDA)
- Thalassemia
- Sideroblastic anemia
- Lead poisoning
- Anemia of chronic disease (ACD)
- Normocytic (MCV 80-100 fL)
- Nonhemolytic (normal or low reticulocyte count)
- Early IDA
- ACD
- Aplastic anemia
- Chronic kidney disease
- Hemolytic (high reticulocyte count)
- Intrinsic
- Sickle cell disease
- Thalassemia
- RBC membrane defects-Hereditary Spherocytosis
- RBC enzyme defect-G6PD Deficiency
- Paroxysmal Nocturnal Hemoglobinuria
- Extrinsic
- Immunohemolytic anemia
- Microangiopathic hemolytic anemia
- Macroangiopathic
- Parasites and other infections
- Intrinsic
- Nonhemolytic (normal or low reticulocyte count)
- Macrocytic (MCV >100 fL)
- Megaloblastic
- Folate deficiency
- Vitamin B12 deficiency
- Orotic aciduria
- Non-megaloblastic
- Liver disease
- Alcoholism
- Diamond-Blackfan anemia
- Megaloblastic
Microcytic Anemia
IDA can be caused by several reasons, loss of blood (menstruation or GI bleed), absorption issues (gastric bypass or celiac disease), or there may be an increased demand as seen in pregnancy. These all can lead to decreased heme synthesis.
Iron is absorbed in the duodenum. If it is heme iron (meat), it is easily absorbed. Regular iron (Fe3+) is not absorbed as well, therefore it is converted to Fe2+ by duodenal cytochrome B. The Fe2+ is then taken into the cell via DMT-1 and is either stored as Ferritin (this is how most iron is stored in the body) or will leave by Ferroportin. Once it leaves the cell it is changed back to Fe3+ and bound to Transferrin (this is how iron is moved in blood). If the body has enough iron, the liver will make Hepcidin, which blocks the Ferroportin. Iron is taken to the bone marrow and the erythroid precursors, which have a receptor for the Transferrin-Fe3+ complex. If the iron is to be stored in the bone marrow, then it is stored as Hemosiderin. The free form of iron is toxic to the body.
Patients with IDA can present with fatigue, SOB, pallor, exercise intolerance, and pica (eating non-foods like dirt-think of it as taking in anything that may have iron). If severe enough, can present with Plummer-Vinson syndrome: microcytic anemia, esophageal webs, and dysphagia.
Lab studies:
Complete Blood Count: will have low amounts of RBC, hemoglobin and hematocrit, and MCV. However, it will have a high Red Blood Cell Distribution Width (RDW), the RDW provides the variation of sizes. Which means, a high RDW, means the RBC sizes vary a lot.
Peripheral Blood Smear: Hypochromic, microcytic RBC, anisocytosis
| Study | Measures | Results | Reference Range |
| Serum iron | Transferrin-iron complex | Decreased | M-65-198 ug/dL
F-26-170 ug/dL |
| Total Iron Binding Capacity (TIBC) | Transferrin only | Increased | 240-450 ug/dL |
| Percent saturation | Iron saturation of transferrin | Decreased | 33% (20-50%) |
| Ferritin | Stores of iron | Decreased | M-20-250 ng/mL
F-10-120 mg/mL |
| Transferrin receptor protein (sTfR) | Erythroid precursor receptor | Increased | 1.8-4.6 mg/L |
| Hepcidin | Levels of hepcidin | Decreased | M-29-254 ng/ml
F-17-286 ng/ml |
Treatment for IDA is oral iron supplementation, there is intravenous iron, but that is not often give. Another option is to give erythropoietin or EPO. This directly stimulates the bone marrow to make RBCs, but this is typically reserved for the severely ill or diabetics/kidney disease. Don’t pull a Lance Armstrong.
Thalassemia is a disease of decreased globin chain synthesis. Because there is decreased globin chain synthesis, there is decreased hemoglobin, and the end result is a microcytic anemia. Thalassemia is an inherited disorder and like sickle cell disease is thought to be protected against malaria. Thalassemia is divided into α-thalassemia (decreased α globin synthesis) and β-thalassemia (decreased β globin synthesis). As previously discussed, normal hemoglobin (HbA, α2β2) is made of two α chains and two β chains. There are two other less significant types of hemoglobin, HbF (α2γ2) and HbA2 (α2δ2).
There are a two genes with four alleles that make the α globin chain (α/α;α/α). An α-thalassemia results when one or more of these genes are deleted.
- Silent carrier (α/-;α/α)
- Carrier
- α-thalassemia trait
- (α/-;α/-)-trans deletion, more likely to occur in Africans, including African Americans
- (-/-;α/α)-cis deletion, more likely to occur in Asians. This is also associated with an increased risk of severe thalassemia in offspring.
- Asymptomatic – no significant hemolysis
- Hypochromic, microcytic anemia & target cells
- HbH Disease (α/-;-/-)
- β chains for tetramers called HbH tetramers
- HbH tetramer damage RBCs
- Moderate hemolyltic anemia
- Hydrops Fetalis (-/-;-/-)
- HbBarts (γ chain tetramer), damage to RBCs
- Lethal in utero
The β globin is usually due to a mutation of the gene (vs deletion in α-thalassemia). There is one gene with two alleles for the β globin. Mutations can result in the absence (β0) or diminished (β+) production of the β globin chains. Because there are only two alleles, there is only β-thalassemia major and β-thalassemia minor.
- β-thalassemia minor (β/β+ or β/β0)
- Asymptomatic
- No significant hemolysis
- Hypochromic microcytic anemia & target cells
- Increased HbA2
- β-thalassemia major (β+/β+ or β+/β0 or β0/β0)
- Unpaired α globin chains precipitate and damage RBC membrane
- Presents @ 6-9 mo as failure to thrive
- Hypochromic microcytes-severe hemolytic anemia
- Jaundice
- Ineffective erythropoiesis-extravascular hemolysis (spleen)
- Extramedullary hematopoiesis, results in hepatosplenomegaly
- Increased EPO, with massive erythroid hyperplasia and bone marrow expansion, results in skeletal deformities (crewcut appearance on x-ray)
- Suppression of hepcidin and an increase in iron absorption
- Increased HbF (γ chains are made to compensate lack of β chains)
- Risk of aplastic anemia with parvo B19 infection
Because of the need for regular transfusions, β-thalassemia major patients can develop iron overload, which can cause hemochromatosis.
Sideroblastic anemia is due to congenital and acquired problems with heme synthesis. All result in defects that cannot incorporate iron into the heme molecule. Through a series of reactions, protoporphyrin is produced, an enzyme called ferrochelatase incorporates iron is then transferred into the protoporphyrin to make the heme molecule. This occurs in the mitochondria. If there is a failure to make the protoporphyrin correctly, this can lead to an abnormal accumulation of iron in immature RBCs called ringed sideroblasts. The iron that is seen is found inside the mitochondria.
Congenital defects are most commonly caused by loss of function of the enzyme that performs the first step in the series of reactions. The enzyme that causes the defect is, aminolevulinate synthase 2 (ALAS), the disease is called X-linked sideroblastic anemia. Due to this loss of function, there is an iron overload in the body, increase in ferritin in the blood, and an increase in transferrin saturation. All this leads to cirrhosis of the liver.
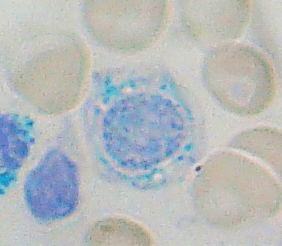
There are various causes of acquired sideroblastic anemia:
- Myelodysplastic neoplasms
- Chromosomes
- Trisomy 8
- Deletion of 5 or 7
- Copper deficiency
- Excess alcohol
- Medications
- Isoniazid (TB drug)
- Chloramphenicol
- Linezolid
- Penicillamine
- Busalfan
Lab studies:
CBC: microcytic hypochromic anemia, decreased reticulocytes, along with the abnormal iron studies mentioned above.
Confirmatory diagnosis is made with bone marrow smears showing the ringed sideroblasts.

Treatment includes removal or addressing the acquired cause and vitamin B6.
Lead poisoning is not very prevalent, but may be seen in those who spend a significant amount of time at the range. Other than anemia, lead toxicity causes:
- Developmental (children)
- Decreased IQ
- Decreased hearing
- Decreased growth
- Impaired PNS
- Altered calcium homeostasis
- Altered vitamin D metabolism
- Decreased nerve conduction velocity
- Colic
- Kidney failure
- Encephalopathy
- Death
Most of the effects of lead toxicity is neurological (peripheral neuropathies). Lead toxicity can cause affect RBCs by causing a sideroblastic anemia as described above. It does this by inhibiting ferrochelatase, which is the last enzyme in the production of heme. It can also inhibit δ-aminolevulinic acid dehydratase.

As mentioned above, the treatment for a sideroblastic anemia is removal of the offending agent, in this case, lead.
Last in our microcytic anemias is anemia of chronic disease (ACD). It is last, because you may see ACD as a microcytic or normocytic anemia. This can be complicated without an understanding of biochemistry. According to Robbins & Cotran Pathologic Basis of Disease ACD is perhaps the most common cause of anemia in hospitalized patients. Chronic disease in this topic is neoplasms, infections, and autoimmune disease. An increased inflammatory state is normal in chronic disease. This results in low serum iron, reduced TIBC, and increased iron stores in macrophages. During chronic disease the liver makes many inflammatory mediators, in this case the most problematic in interleukin-6 (IL-6), which causes an increase in hepcidin production. Recall that hepcidin inhibits ferroportin. This leaves little iron to make hemoglobin. IL-6 can also inhibit EPO, causing further issues with RBC production. So, why does this happen? A couple of possibilities, one-bacteria need iron to live, therefore if the body restricts its availability, it plays in our favor. This is not necessarily a bad thing in the acute illness. Second-macrophages can use iron to kill certain bacteria like H. influenzae.
Lab studies:
Complete Blood Count: may have low amounts of RBC, hemoglobin and hematocrit, and MCV.
Iron studies: decreased serum iron, TIBC, percent saturation. Increased ferritin and hepcidin.
Peripheral Blood Smear: normocytic or microcytic, hypochromic or normochromic.
While a medic is not going to do a bone marrow smear or aspirate, you will look smart if someone asks: with Prussian Blue Stain, there will be ample iron stores and this is how you can differentiate between IDA and ACD.
The definitive treatment is to treat the underlying issue. However, there are times when iron supplementation, EPO, or even blood transfusions may be necessary.
See: Pathogenesis and treatment of anaemia of chronic disease
Normocytic Anemia
Normocytic anemia can be divided into hemolytic or non-hemolytic. Nonhemolytic has a normal or low reticulocyte count, where hemolytic has a high reticulocyte count. IDA and ACD has been previously discussed, the obvious difference in the MCV, where normocytic is within the normal range.
Nonhemolytic
Aplastic anemia (AA) refers to a significant reduction of all blood cells (RBCs, PLTs, WBCs), or pancytopenia. This is usually caused by a autoimmune disease, but inherited and acquired abnormalities can also cause AA. This is a rare issue to see in military members, if you do come across it, it is important to be aware of the dangers. They are at risk for infections and bleeding. Patients can present with weakness, pallor, and shortness of breath. This can be extremely serious and may require you to perform a blood transfusion if in a PFC scenario.
Acquired causes:
- Idiopathic
- Stem cell defects
- Immune mediated
Chemical agents:
- Dose related
- Alkylating agents
- Antimetabolites
- Benzene
- Chloramphenicol
- Idiosyncratic
- Chloramphenicol
- Phenylbutazone
- Carbamazepine
- Gold salts
- Penicillamine
Physical agents:
- Whole Body Irradiation
- Infections-EBV, CMV, rubella, viral hepatitis
Inherited disorders:
- Fanconi Anemia
- Telomerase defects
While it is not fully understood and most likely multifactorial, two major etiologies are identified. An extrinsic, immune mediated that suppresses the bone marrow and an intrinsic, which is an issue with the stem cells.
See: How I treat acquired aplastic anemia (2017)
See: Guidelines for the diagnosis and management of adult aplastic anaemia (2015)
Chronic kidney disease that results in anemia is secondary to the decreased EPO release. Recall that EPO or erythropoietin is released by the kidney in response to low oxygen content. EPO travels to the bone marrow where it stimulates RBC proliferation and maturity. In chronic kidney the loss of kidney function, decreases the amount of EPO that is made and therefore released.
Treatment includes administration of erythropoiesis stimulating agents (ESAs), which include EPO (Epoetin Alpha), but also one that has a much longer half-life: Darbepoeitin (NESP). The hemoglobin and hematocrit goal is 11 g/dL and 33-36%. This is due to a higher risk if MIs with hematocrit >40%.
See: Anemia of Chronic Kidney Disease (2008)
Hemolytic anemia
Hemolytic anemia is caused by intrinsic and extrinsic issues. The intrinsic causes should be somewhat familiar, they include:
- Sickle cell disease
- Thalassemia
- RBC membrane defects-Hereditary Spherocytosis
- RBC enzyme defect-G6PD Deficiency
- Paroxysmal Nocturnal Hemoglobinuria
General features of hemolytic anemias:
- Destruction of RBC before normal life span (100-120 days)
- Jaundice (conjunctiva/skin)-from increase in indirect bilirubin,
- Increase in lactate dehydrogenase (LDH)-found in large numbers in RBCs
- Increase Reticulocyte Index (RI)
Complete Blood Count:
- Usually normocytic
- Normochromic anemia (can have macrocytosis from extensive reticulocytosis also hypochromia and microcytosis for thalassemia)
PBS: Polychromasia from reticulocytosis, schistocytes, and other disease specific findings
Labs:
- Serum: increased indirect bilirubin and LDH, decreased haptoglobin.
- Urine: hemogloinuria and hemosiderinuria
Sickle cell disease (SCD) is relatively common in the US. SCD is is autosomal recessive disease and sickle cell trait (SCT) is autosomal dominant. It was thought SCT was preventive against malaria, SCD and SCT is most commonly found in blacks. SCD is caused by the replacement of an amino acid (glutamate) to another (valine) at position six on the β-chain. Because of this, it causes the RBC to be hydrophobic when it is deoxygenated. This hydrophobic nature, causes the RBC to “sickle” and become sticky. When these RBCs become sticky and sickle, they can cause vaso-occlusive crises. This can be painful and cause increased risk of infarction and infections. Other complications that are associated with SCD:
- Aplastic crisis with Parvovirus B19 infection
- Autosplenectomy – the sickle cells get stuck in spleen, which then infarcts
- Death by encapsulated organisms is most common in children
- Fibrosis and atrophy of the spleen
- Folic acid deficiency
- Salmonella osteomyelitis
- Renal papillary necrosis
- Avascular necrosis of hip
- Acute Chest Syndrome – vaso-occlusion in pulmonary microcirculation; precipitated by pneumonia. This is the most common cause of death in adults with SCD.
- Priapism
- Extramedullary erythropoiesis
- Elevated reticulocyte index (RI) reflecting adequate marrow response
- Low RI suggests inadequate response to anemia and lack of erythropoiesis
Complete Blood Count: there will be decreased amounts of RBC, hemoglobin and hematocrit, there will be an increased reticulocyte count.
Labs: an increase in indirect bilirubin and LDH with a low haptoglobin
Urine: it may show a microalbuminuria and macroalbuminuria
Every day treatment includes hydroxyurea and hydration. In the acute setting, you can treat with IV infusion of 0.9% normal saline, supplemental oxygen, and analgesics (ketamine and opioids).

By The National Heart, Lung, and Blood Institute (NHLBI) – http://www.nhlbi.nih.gov/health/health-topics/topics/sca/, Public Domain, https://commons.wikimedia.org/w/index.php?curid=19198765

Hereditary Spherocytosis (HS) is a RBC membrane defect issue. It is an autosomal dominant disease, meaning if one of the parents carries the gene and passes it to the offspring, he/she will have the disease. The highest prevalence is in northern Europe.
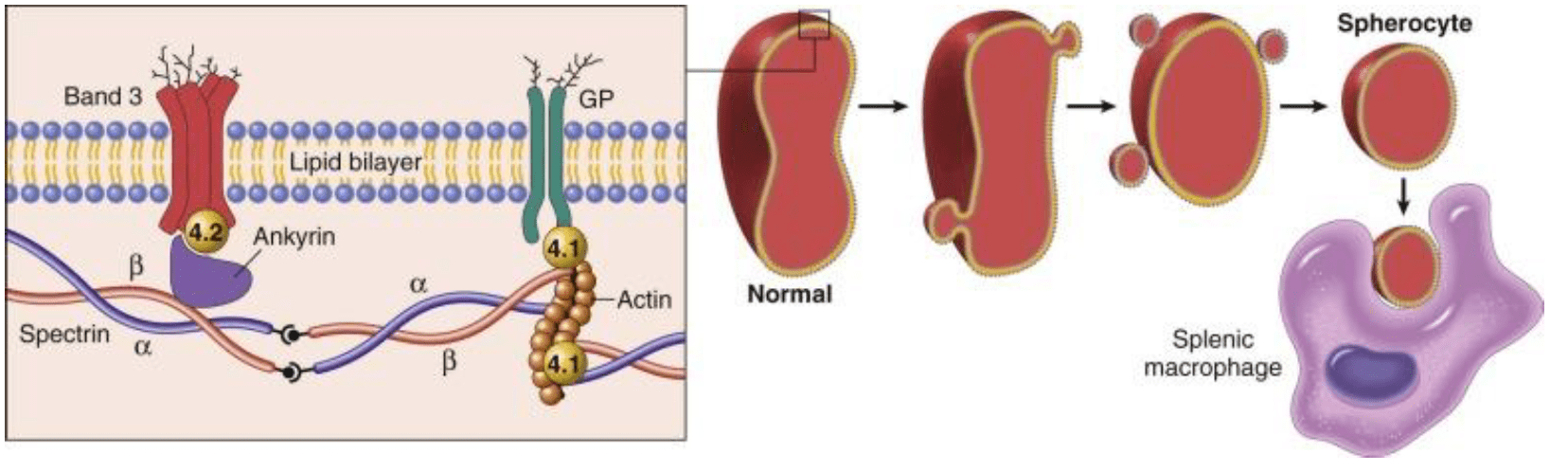
The mutation for HS is most commonly found in Ankyrin, Spectrin, Band 3 and Band 4.2. These proteins are responsible for the RBC maintaining its biconcave shape. Several proteins contribute to the RBCs skeleton. Because of the RBC has a defective skeleton, the RBC has a distinctive appearance, appropriately called a spherocyte. The membrane blebs lost over time and caused a loss of central pallor. Eventually, the RBC becomes spherical and fragile

Because of these abnormal shaped cells, when they reach the spleen, they are destroyed. This splenic destruction causes the anemia. You may see Howell-Jolly bodies, these are pieces of nuclear remnants that are normally degraded by spleen. This can be a sign of markedly decreased splenic function.
A patient could present with the following:
- Mild to moderate hemolytic anemia
- Jaundice
- Splenomegaly
- Pigment gallstone
- Folic acid deficiency
- Aplastic crisis can occur with a Parvo B19 infection
- Hemolytic crisis can occur with certain drugs or infections affecting the BM
Complete Blood Count: Increased Mean Corpuscular Hemoglobin Concentration (MCHC), increased RDW
PBS: Spherocytes, Howell-Jolly bodies
Labs: Osmotic Fragility Test (incubate in hypotonic salt solution; spherocytes show ↑ in osmotic fragility compared to normal red blood cells), flow cytometry, or molecular testing finding the underlying mutation.
Treatment for HS is a Splenectomy and treat aplastic crisis/hemolytic crisis with blood transfusions
Glucose-6-phosphate dehydrogenase deficiency (G6PD deficiency) should be somewhat familiar to military medics, it is a test commonly ordered for physicals. G6PD Deficiency is an issue with enzymes in RBC. It’s an X-linked disease, meaning it is mostly males who are affected and is seen in people of African and Mediterranean decent.
The G6PD enzyme maintains adequate intracellular “reduced glutathione,” which allows for protection of the RBC against oxidative damage. Without this enzyme functioning it RBCs are more susceptible to oxidative damage.

Patients are usually asymptomatic, but there is episodic hemolytic anemia without chronic signs or symptoms of chronic hemolysis. The hemolytic anemia occurs only with certain exposures:
- Infections (release of free radicals)
- Foods e.g. fava beans
- Drugs e.g. antimalarial meds, antibiotics (sulfonamides, dapsone), anti-inflammatories
After the exposure, symptoms will begin to appear 1-2 days later. These signs and symptoms are typical of a hemolytic anemia, with jaundice or red-brown urine (from hemoglobinuria and hemosiderinuria)
CBC: mild normocytic anemia
PBS: bite cells and Heinz bodies (these represent denatured globin chains)

Labs: measure G6PD enzyme levels
Treatment for G6PD Deficiency is for the most part, preventative; stop offending agent/exposure; provide supportive care since disease is usually self-limiting. This is why we test for it often on physicals.
Paroxysmal Nocturnal Hemoglobinuria (PNH) is out last intrinsic cause of hemolytic normocytic anemia. It is slightly more complicated and indepth than some of the other anemias. This is caused by acquired somatic mutations. These mutations inactivate X-linked PhosphatidylInositol glycan complementation Group A (PIGA) which affect the hematopoietic stem cells, hence all subsequent clones are affected (red blood cells, white blood cells and platelets).

PNH blood cells are deficient in three GPI-linked proteins that regulate complement activity:
- Decay-accelerating factor, or CD55
- Membrane inhibitor of reactive lysis, or CD59
- CD59, is a potent inhibitor of C3 convertase that prevents the spontaneous activation of the alternative complement pathway
- C8 binding protein
Patients present with paroxysmal-hemolysis it can be recurrent or episodic, however, it can be chronic as well. The nocturnal-hemolysis occurs at night/during sleep, because at night, the body’s pH lowers and becomes more acidotic. Patients wake up and their urine is red-brown. Other complaints, Smooth muscle spasm-abdominal pain, difficulty swallowing (from esophageal spasm) and thrombosis, there is an increased risk of venous thrombosis. Here is the issue, chronic hemolysis can occur at any time. The hemolysis is intravascular.
Complete Blood Count: Normocytic anemia with increased reticulocyte index.
Urine: hemoglobinuria and hemosiderinuria.
Labs: increased indirect bilirubin, LDH, decreased haptoglobin.
Treatment for PNH is eculizumab, a monoclonal antibody to prevent conversion of C5 to C5a. They may also be prescribed immunosuppressive therapy. In severe cases, a bone marrow transplant is needed.

Leave a comment