
Nephritic syndrome is typically characterized by inflammation of the glomeruli and presents with hematuria, red cell casts, azotemia, oliguria, proteinuria, and hypertension.
Acute proliferative glomerulonephritis is the result of a post infectious state (Postreptococcal). There is a diffuse proliferation of glomerular cells and influx of WBCs. The lesions are the result of immune complex deposition and can be exogenous (Poststreptococcal) or endogenous (SLE).
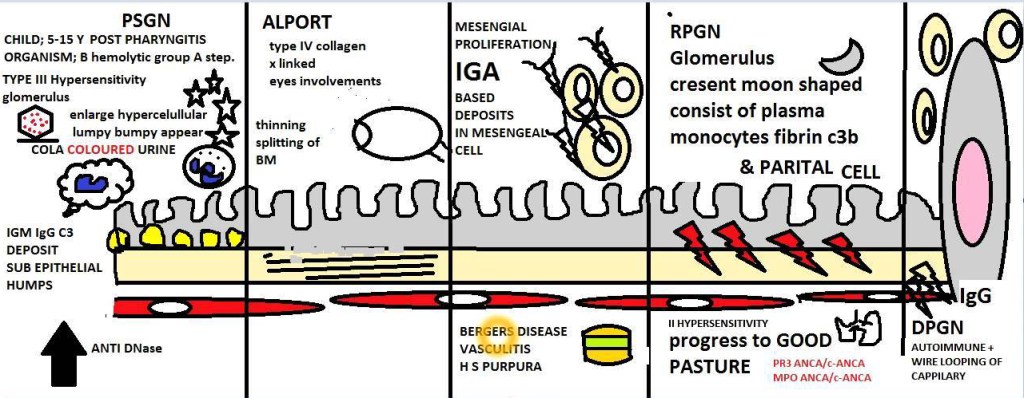
Poststreptococcal Glomerulonephritis (PSGN) used to be somewhat common before antibiotics, but since it has decreased substantially. It can appear 1-4 week post infection streptococcal infection (pharynx/impetigo). The age is usually between 6-10 years of age. PSGN is caused by immune complex containing streptococcal antigens and specific antibodies (IgG). Group A β-Hemolytic Streptococci (GABHS) is the primary offender. The rise in titers and decreasing level of complement, indicating complement activation.
The main component responsible for PSGN is the streptococcal pyogenic exotoxin B (SpeB). However, it is not the cause in every case. SpeB activates complement and is the localized “hump-like” deposits. For a reason not completely understood, antigen-antibody complexes dissociate, cross the glomerular basement membrane and the reform on the subepithelium.

The clinical course for PSGN in children will present as sudden onset of fever, malaise, nausea, oliguria, and hematuria 1-2 weeks post infection. The examination of the urine will have red cell casts and mild proteinuria. A patient may have periorbital edema and mild to moderate hypertension. In a adults, there tends to be onset of hypertension, edema, and a rise in the blood urea nitrogen (BUN). It is important to draw antistreptococcal antibody titers to evaluate for the presence of this condition in a subclinical illness.
Most pediatric patients will recover with conservative treatment and without any decrease in long term renal function. A very small amount of folks (~1%) do not improve and will become severely oliguric and will develop a rapidly progressive glomerulonephritis and some may develop a chronic glomerulonephritis.

The adult patient tends to have a more difficult course. While most (60%) will recover, the rest develop persistent proteinuria, hematuria, and hypertension. Some will then clear the issue, while others proceed to a rapidly progressive glomerulonephritis.
Other forms of the non-PSGN include staphylococcal, pneumococcal, HIV, Hepatitis C, EBV, varicella, malaria, and toxoplasmosis. This can be identified by granular immunofluorescent deposits and subepithelial bumps. In the staphylococcal form, the antibody is IgA.
The rapidly progressive glomerulonephritis (RPGN) is associated with severe glomerular injury, but is not a specific etiologic form of glomerulonephritis. RPGN is characterized by a rapid and progressive loss of renal function and has severe oliguria and signs of nephritic syndrome. If not treated, this will progress to acute renal failure and death within weeks. The most common histologic finding is the presence of crescents in the glomeruli (crescentic glomerulonephritis). There is a proliferation of the parietal epithelial cells lining Bowman’s capsule with infiltration of macrophages and monocytes.

Type I or anti-GBM antibody is mediated by linear deposits of IgG and C3 in the GBM. THe antigen common to GBM and alveoli is a peptide (α3) within the type IV collagen. The trigger for the formation of these antibodies is unclear at this time. Treatment is plasmapheresis to remove the IgG.
Type II or immune complex deposition occurs in many different types of diseases, but common is the granular pattern of staining and immunofluorescence. Here, there will be proliferation and influx of WBCs in the glomerular tuft in addition of crescent formation. Typically the treatment is fixing the underlying disease.
Type III or pauci-immune, there is a lack of detectable anti-GBM antibodies or immune complexes. Most have ANCA circulating that produce cytoplasmic (c) or perinuclear (p) staining patterns.
RPGN types:
- Type I (anti-GBM antibody)
- Renal limited
- Goodpasture syndrome
- Type II (immune complex)
- Idiopathic
- Postinfectious glomerulonephritis
- SLE nephritis
- Henoch-Schönlein purpura
- IgA nephropathy
- Type III (pauci-immune)
- ANCA associated
- Idiopathic
- Granulomatosis with polyangiitis
- Microscopic polyangiitis
The renal manifestations of all forms of crescentic glomerulonephritis include oliguria, proteinuria (almost reaching nephrotic range), and hypertension. Goodpasture will have hemoptysis too. Labs test to help confirm this diagnosis include anti-GMB antibodies, antinuclear antibodies, and ANCAs.

Leave a comment